

JIS T 81001-5-1に準拠した医療機器のセキュリティ対策
更新:2024年4月17日
医療機器の基本要件基準は、2023年3月9日厚生労働省告示第67号により第12条第3項「サイバーセキュリティに関する要求事項」が新設され、2023年4月1日より運用が開始されました。移行期間の1年は2024年3月31日をもって終了し、今後、機器メーカーは、機器の承認申請に際して新規制への適合性を証明する必要があり、基本要件基準に適合しない医療機器の販売および製造などを行うことはできません。
ユビキタスAIでは、JIS T 81001-5-1に準拠した医療機器のサイバーセキュリティ対策として、医療機器セキュリティ検証サービスを提供しております。「コンサルティング」「脆弱性スキャン」「ファジングテスト」「ペネトレーションテスト」等、ご要望に応じたサービスメニューのご対応が可能です。まずはお気軽にお問合せください。
IoT機器セキュリティ検証
サービスの詳細を見る お気軽相談をする
医療機器のサイバーセキュリティに求められる対策とユビキタスAIの提供する検証サービス
IMDRFガイダンス
世界中で急増する医療機関におけるセキュリティインシデント対策として、2020年に国際医療機器規制当局フォーラム(IMDRF)が 「医療機器に関するサイバーセキュリティ対策の原則及び実践」(IMDRFガイダンス)を発行しました。
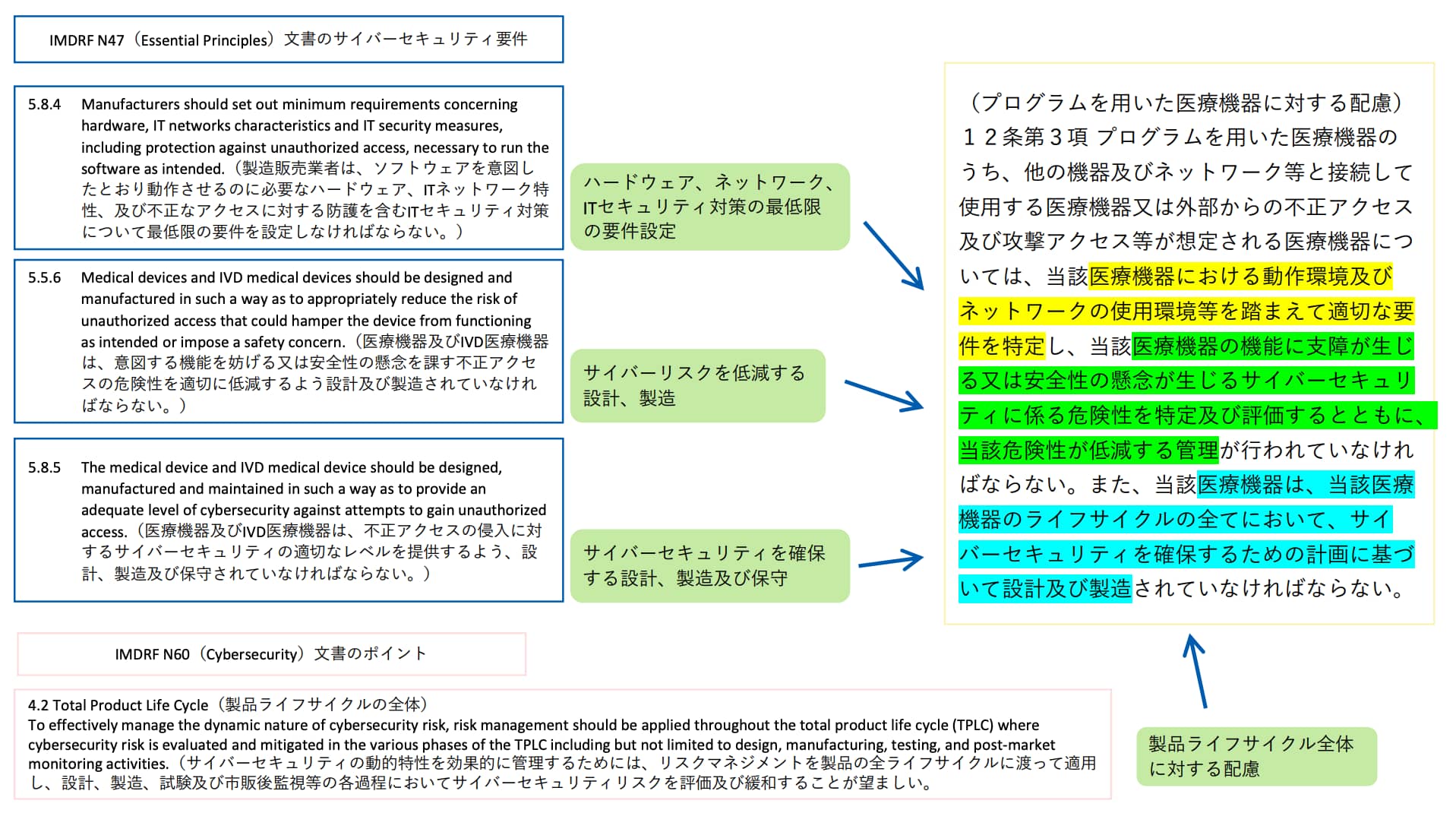
出典:https://www.mhlw.go.jp/content/11121000/001037562.pdf
医療機器のサイバーセキュリティ導入に関する手引書
国内で医療機器におけるサイバーセキュリティのリスクを評価し、リスクを管理するためのガイドラインやプロセスを提供する文書です。主に医療機器製造業者向けの製品ライフサイクルにおける要求事項がまとめられています。2021年に「医療機器のサイバーセキュリティ導入に関する手引書」が発行され、2023年にはIMDRF追補ガイダンスの内容を追加して「医療機器のサイバーセキュリティ導入に関する手引書(第2版)」が発行されました。第2版には主に下記が追加されました。
- SBOM(ソフトウェア部品表)の取り扱い
- レガシー医療機器の取り扱い
- 脆弱性に関する修正
- インシデントの対応
JIS T 81001-5-1
医療機器に組み込むソフトウェアを対象としたプロセス規格で、JIS T 2304(医療機器ソフトウェアライフサイクルプロセス規格)に準拠した開発を進める上で実施すべきサイバーセキュリティに関する対策を規定しています。IMDRFガイダンスを導入すべく、厚生労働省より「基本要件基準第12条第3項の適用について」(薬生機審発0331第8号)が通知され、医療機器のサイバーセキュリティの確保について、プログラムを用いた医療機器に関しては「JIS T 81001-5-1」への適合性の確認をもって基本要件基準第12条第3項への適合とすること、プログラムを用いた医療機器は「JIS T 81001-5-1」に準拠したサイバーセキュリティ対策が必要であることが明記されています。
3.基本要件基準第12条第3項の適用・適合性の確認について
(1) プログラムを用いた医療機器の製造販売業者、外国製造医療機器等特例承認取得者又は外国指定高度管理医療機器製造等事業者(以下「製造販売業者等」という。)はこれまでもJIS T 2304によって、医療機器ソフトウェアライフサイクル全体を通じて、適切なリスクマネジメントを実施することにより、医療機器の安全性と基本性能を確保することが求められてきたところであるが、プログラムを用いた医療機器については、これに加えて、JIS T 81001―5―1によって、製品ライフサイクルにおける取組を通じたサイバーセキュリティ対策をより強化し、サイバーセキュリティに関するリスクを許容可能な範囲となるまで低減し、患者への危害の発生及び拡大の防止に繋げる必要があること。
※ JIS T 81001―5―1(ヘルスソフトウェア及びヘルスITシステムの安全、有効性及びセキュリティ―第5―1部:セキュリティ―製品ライフサイクルにおけるアクティビティ)は、医療機器の製造販売業者がJIS T 2304(医療機器ソフトウェアライフサイクルプロセス)に規定する製品ライフサイクルの要求事項に加えて実施するサイバーセキュリティに関する取組を規定している。
(2) JIS T 81001―5―1の他、プログラムを用いた医療機器のサイバーセキュリティの確保について、IEC 81001―5―1等の国際的に用いられている適切な規格等への適合性を確認することをもって基本要件基準第12条第3項への適合を確認したものとして差し支えないこと。なお、承認申請(承認事項一部変更承認申請を含む。以下同じ。)又は認証申請(認証事項一部変更認証申請を含む。以下同じ。)に際しては、それらの規格等を用いることの妥当性を説明すること。
(3) 製造販売業者等は、プログラムを用いた医療機器のサイバーセキュリティの確保の確認及び検証を適切に考慮及び実施する体制を整備し、その適合に関する確認等の実施を適切に記録し保管すること。医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(昭和35年法律第145号)第23条の2の5第7項又は第23条の2の23第4項の規定による調査の調査権者の求めなどに応じて資料を提示し、適切な説明を行わなければならないこと。
(4) 高度管理医療機器又は管理医療機器の承認申請又は認証申請を行う製造販売業者等は、申請に当たり、当該医療機器についてJIS T 81001―5―1等への適合性を示す資料を添付する必要があること。
また、一般医療機器についても同様に適合性を確認する必要があるが、届出の際に資料の添付は要さないこと。
4.経過措置について
本改正で追加される基本要件基準第12条第3項は令和5年4月1日より適用されるが、令和6年3月31日までの間、なお従前の例によることができること。
(1) 改正後の基本要件基準が適用される令和6年3月31日以前に承認若しくは認証を受けた医療機器又は届出された医療機器については、改めて申請・届出を行う必要はないものとする。
ただし、承認認証事項又は届出事項に何らかの変更が生じる等により、令和6年4月1日以降に当該医療機器の承認又は認証事項一部変更申請が必要な場合にあっては、改正後の基本要件基準への適合を確認した上で、改正後の基本要件基準への適合を示す資料を添付すること。
なお、令和6年4月1日以降に製造販売する医療機器は、改正後の基本要件基準への適合を確認した上で、改正後の基本要件基準への適合に関する資料を求めに応じて提示できるようにしておくこと。
令和6年3月31日以前に製造販売された医療機器に関する取扱いについては追って通知するものとする。
(2) 令和6年3月31日以前に承認申請若しくは認証申請又は届出される医療機器については、承認申請又は認証申請若しくは届出時に、改正後の基本要件基準への適合を示す資料を添付する必要はないものとする。
令和6年4月1日以降の取扱いについては、4.(1)のただし書き及びなお書きを準用する。
(3) 令和6年4月1日以降に承認申請若しくは認証申請を行う医療機器については、改正後の基本要件基準への適合を確認した上で、改正後の基本要件基準への適合に関する資料を添付する必要がある。届出を行う医療機器についても、改正後の基本要件基準への適合を確認すること。
出典:https://www.mhlw.go.jp/web/t_doc?dataId=00tc7558&dataType=1&pageNo=1
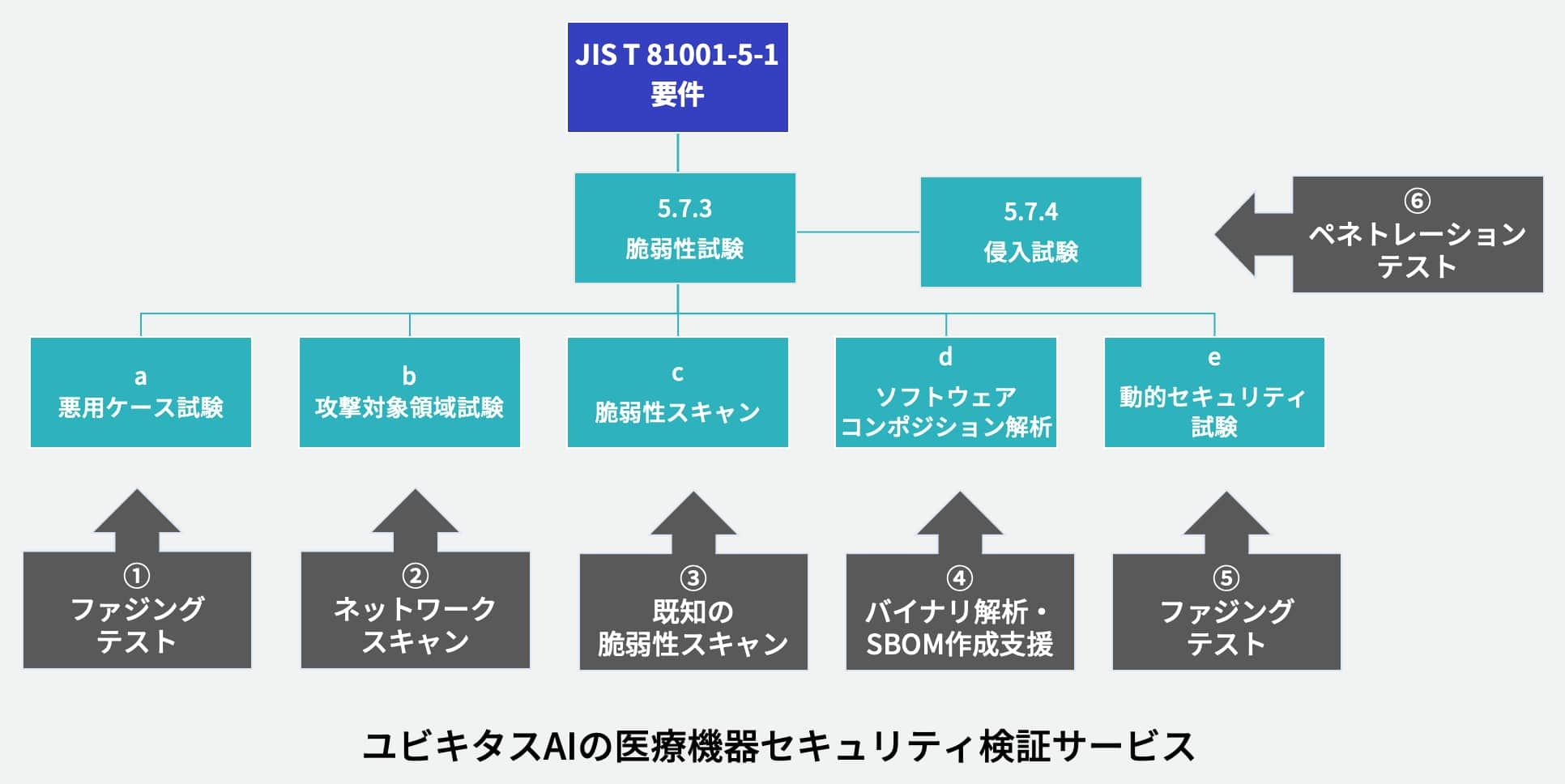
ユビキタスAIの医療機器向けセキュリティ検証サービス内容
- コンサルティングサービス:製品固有の特性に合わせたセキュリティ検証のご提案、検知した脆弱性に対する対応などのご相談に応じます。
- 既知脆弱性の診断:既知の脆弱性が機器に内在しうるかを調べ、実際に悪用可能かを確認します。
- ファジングテスト:製品に搭載されているインタフェースに対して 問題が起こりそうなデータや改変したデータを挿入し、その挙動を確認します。
- ネットワークスキャン:どのポートに対して通信可能か、接続が許可されていない機器やサービスが存在しないかを確認します。
- ネットワークキャプチャ:機器やサービスのネットワークパケットを取得し、平文の通信、暗号強度の低い通信などがないかを確認します。
- ペネトレーションテスト:製品の仕様に基づき、攻撃の可否を確認します。
- レポート:客観性かつ再現性のある検証結果報告書をご提示します。
各国のIoT製品セキュリティ確保のための取り組み: 英国
2024.04.18
ソフトウェアテストの新常識:ファジング入門
2024.04.17

各国のIoT製品セキュリティ確保のための取り組み:欧州
2024.01.26

各国のIoT製品セキュリティ確保のための取り組み:米国
2023.12.18
サプライチェーン攻撃と脆弱性テスト
2023.12.14

セキュリティ規格について
2023.09.01

ファジングとは?
2023.09.01

脆弱性検証―何をどこまで実施すれば良い?
2023.09.01

HEMS機器の脆弱性検証
2023.07.14
ファジングの限界
2023.07.14
